Table of Contents
Overview – Congenital Adrenal Hyperplasia
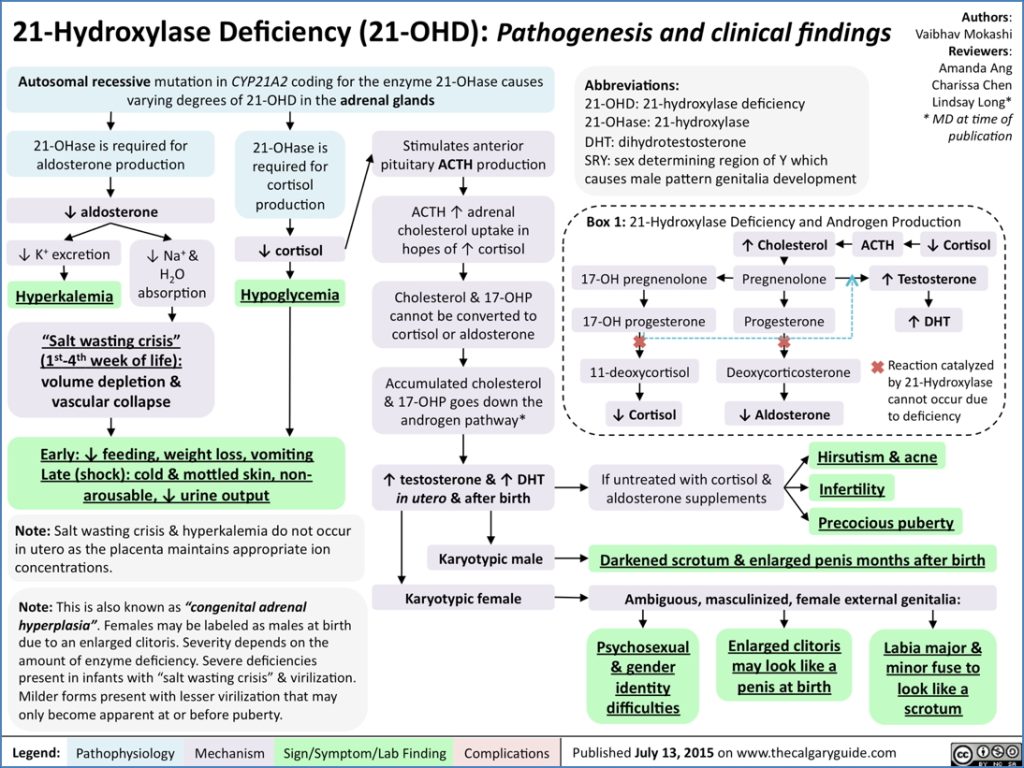
Congenital Adrenal Hyperplasia (CAH) is a group of autosomal recessive disorders affecting adrenal steroidogenesis, with 21-hydroxylase deficiency accounting for over 90% of cases. This enzyme deficiency leads to reduced cortisol and aldosterone synthesis, resulting in compensatory ACTH overproduction and shunting towards androgen synthesis. CAH presents along a spectrum, from life-threatening neonatal salt-wasting crises to ambiguous genitalia and premature puberty. Early diagnosis and hormonal replacement therapy are key to management and preventing long-term complications.
Definition
CAH is a genetic disorder caused by enzymatic defects in adrenal steroid biosynthesis, most commonly due to 21-hydroxylase deficiency, leading to cortisol and aldosterone deficiency with androgen excess.
Aetiology
- Inheritance: Autosomal recessive
- Most common cause: 21-hydroxylase deficiency due to mutations in the CYP21A2 gene
Pathophysiology
- 21-hydroxylase is required for synthesis of both cortisol and aldosterone
- Deficiency leads to:
- ↓ Cortisol → ↑ ACTH (via negative feedback) → adrenal hyperplasia
- ↓ Aldosterone → ↓ Na⁺ reabsorption, ↑ K⁺ retention → hypovolaemia, hyponatraemia, hyperkalaemia
- ↑ 17-hydroxyprogesterone accumulates and is shunted to androgen synthesis → virilisation
Clinical Features
Neonates
- Salt-wasting crisis (common in severe forms):
- Presents in 1st week of life
- Hypovolaemic shock, dehydration, vomiting, hypotension
- Mottled skin, non-arousable
- Ambiguous genitalia in females (enlarged clitoris, labial fusion)
- Normal genitalia in males but may have hyperpigmentation or enlarged genitalia
Older Children / Adolescents
- Signs of androgen excess:
- Females:
- Hirsutism
- Oligomenorrhoea or amenorrhoea
- Acne
- Clitoral hypertrophy
- Males:
- Precocious puberty
- Penile enlargement
- Small testes (due to suppressed gonadotropins)
- Oligospermia
- Females:
- Psychosocial & gender identity issues (in severe virilisation)
Investigations
- Serum electrolytes:
- Hyponatraemia
- Hyperkalaemia
- Serum 17-hydroxyprogesterone: ↑↑
- ACTH: ↑
- Cortisol and aldosterone: ↓
- Karyotyping (for ambiguous genitalia)
- Ultrasound: Adrenal enlargement, internal genitalia assessment
Management
- Glucocorticoid replacement (e.g. hydrocortisone): suppresses ACTH → ↓ androgen overproduction
- Mineralocorticoid replacement (e.g. fludrocortisone) if salt-wasting
- Salt supplementation in neonates
- Surgical management of ambiguous genitalia (controversial; requires multidisciplinary approach)
- Lifelong follow-up for hormone adjustment, fertility, and psychological support
Complications
- Adrenal crisis (life-threatening salt-wasting)
- Precocious puberty
- Infertility
- Short stature (due to premature epiphyseal closure)
- Psychological distress due to gender ambiguity
Summary – Congenital Adrenal Hyperplasia
Congenital Adrenal Hyperplasia (CAH), most often due to 21-hydroxylase deficiency, leads to cortisol and aldosterone deficiency with androgen excess. It may present in infancy with life-threatening salt-wasting or later with virilisation and puberty disorders. Early diagnosis and hormone replacement therapy are vital for long-term outcomes. For related content, visit our Endocrine Overview.