Table of Contents
Overview – Multiple Endocrine Neoplasia
Multiple Endocrine Neoplasia (MEN) refers to a group of rare, inherited disorders characterised by the development of multiple tumours in endocrine glands such as the parathyroid, pituitary, pancreas, thyroid, and adrenal medulla. These syndromes significantly increase the risk of both benign and malignant tumours, often with early onset and aggressive progression. MEN syndromes are divided into subtypes (MEN1, MEN2a, MEN2b) based on their genetic mutation and characteristic tumour pattern.
Definition
- A group of autosomal dominant genetic disorders
- Characterised by neoplasia (tumours) in two or more endocrine glands
- Tumours may be benign or malignant
Aetiology
- Inherited mutations in tumour suppressor genes
- Each subtype involves a different gene and clinical spectrum:
- MEN1: MEN1 gene mutation on chromosome 11
- MEN2a & MEN2b: RET proto-oncogene mutation on chromosome 10
Classifications
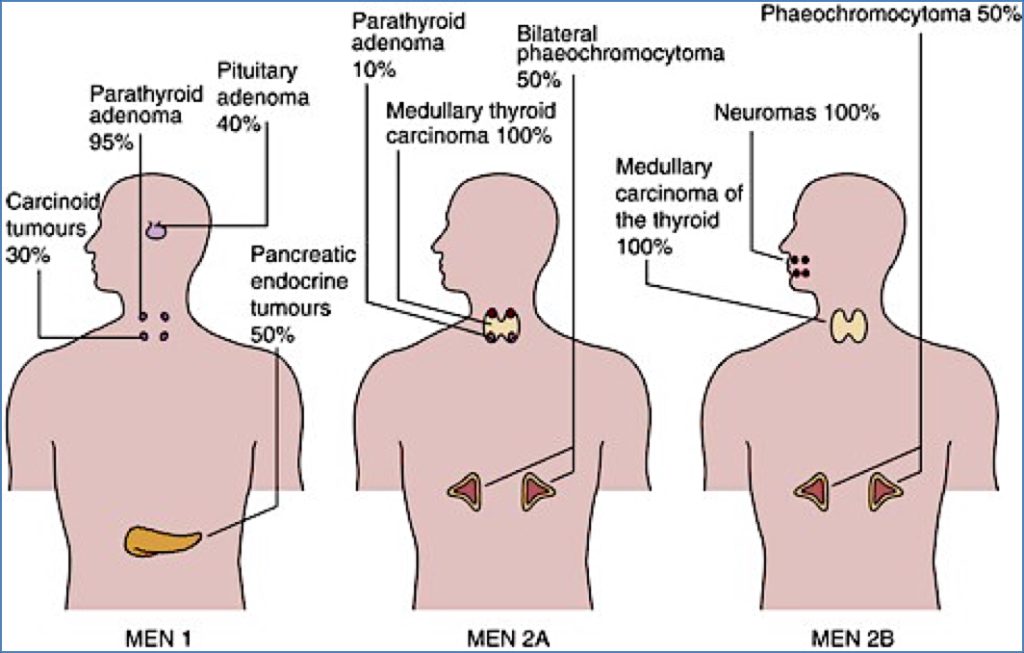
MEN1 – Wermer’s Syndrome
- Gene: MEN1 (tumour suppressor, chromosome 11)
- Organs Involved:
- Pituitary
- Parathyroid
- Pancreas
- Clinical Features:
- 60% of patients have ≥2 of the following:
- Pituitary adenomas (e.g. prolactinoma → amenorrhoea/galactorrhoea, acromegaly, Cushing’s)
- Parathyroid adenomas → primary hyperparathyroidism
- Pancreatic endocrine tumours:
- Gastrinoma → Zollinger-Ellison Syndrome (severe peptic ulcers)
- Insulinoma → recurrent hypoglycaemia
- VIPoma → watery diarrhoea, hypokalaemia
- 60% of patients have ≥2 of the following:
MEN2a – Sipple’s Syndrome
- Gene: RET proto-oncogene (chromosome 10)
- Organs Involved:
- Thyroid
- Parathyroid
- Adrenal Medulla
- Clinical Features:
- 100% of patients → Medullary thyroid carcinoma
- 50% → Phaeochromocytoma → episodic hypertension
- 30% → Parathyroid hyperplasia → hyperparathyroidism
MEN2b
- Gene: RET proto-oncogene (chromosome 10)
- Organs Involved:
- Thyroid
- Adrenal Medulla
- Clinical Features:
- 100% → Medullary thyroid carcinoma (often aggressive and early onset)
- 50% → Phaeochromocytoma
- Additional:
- Mucosal neuromas (e.g. lips, tongue)
- Marfanoid habitus (tall stature, long limbs, joint laxity)
Clinical Features Summary
| Feature | MEN1 | MEN2a | MEN2b |
|---|---|---|---|
| Thyroid carcinoma | No | Medullary (100%) | Medullary (100%) |
| Parathyroid involvement | Hyperparathyroidism | Hyperparathyroidism (30%) | Rare |
| Pituitary tumours | Common | No | No |
| Pancreatic tumours | Gastrinoma, Insulinoma, etc. | No | No |
| Phaeochromocytoma | No | 50% | 50% |
| Other | — | — | Mucosal neuromas, marfanoid |
Management
- Genetic Testing for family members
- Regular screening of endocrine function and tumour markers
- Surgical resection for identified tumours
- Prophylactic thyroidectomy (especially in MEN2 with RET mutation)
- Hormonal replacement therapy if glands are removed or hypofunctional
Summary – Multiple Endocrine Neoplasia
Multiple Endocrine Neoplasia (MEN) syndromes are inherited conditions predisposing individuals to develop multiple endocrine tumours. MEN1 involves the parathyroid, pituitary, and pancreas; MEN2a and MEN2b involve the thyroid (medullary carcinoma) and adrenal medulla (phaeochromocytoma), with MEN2b also presenting with mucosal neuromas and marfanoid features. Early diagnosis, screening, and surgical management are essential. For a broader context, see our Endocrine Overview page.